
Application of Good Clinical Practice at UW-Madison
This document describes Good Clinical Practice (GCP), provides investigators, sponsor-investigators, and study teams with a decision tree to determine if their research must follow the standards described in GCP, and includes detailed guidance about what it means to follow GCP.
NOTE: In July 2025, ICH GCP E6 (R3) was issued as an update to the ICH GCP E6 (R2) Integrated Addendum. The below information still refers to R2.
What is Good Clinical Practice (GCP)? The term GCP is used interchangeably in reference to both a broad concept and a single document.
GCP is often used broadly to describe what are considered best practices when conducting clinical research. These practices set the standards for designing, conducting, performance monitoring, auditing, recording, analysis, and reporting of clinical research that involve human subjects. Compliance with this standard helps to provide public assurance that the rights, safety, and welfare of human subjects in research are protected; that the research is consistent with the principles that have their origin in the Declaration of Helsinki; and that the data collected are reliable and credible.
The term GCP is also used more narrowly to describe the International Council for Harmonisation (ICH) GCP E6 document itself. GCP describes the unified, international standard for the European Union, Japan, the United States, Canada, and Switzerland to facilitate the mutual acceptance of data from clinical trials by the regulatory authorities in these jurisdictions.
The applicability and application of GCP differs based on the specifics of the study. Use the decision tree below to help determine if GCP applies to your study. Click on the links below the tree* for additional information contained within this document, as well as other external resources.
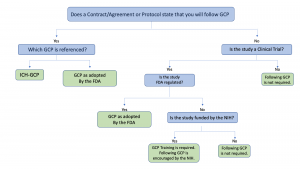
*Links to assist with answering questions:
- Is the study a clinical trial?
- Is the study FDA-regulated?
- UW-Madison GCP Training Requirements
How are ICH GCP and GCP as adopted by the FDA related?
ICH GCP Guidelines should be followed when generating clinical trial data that are intended to be submitted to international regulatory authorities. In protocols or contracts/agreements, when the international standards are to be followed, it is referred to as ICH GCP.
The United States Food and Drug Administration (FDA) adopted the ICH GCP E6 (R2) Integrated Addendum as guidance in March 2018. FDA regulated clinical trials must follow the aspects of GCP that are consistent with the applicable FDA regulations. The aspects of GCP that are not otherwise described in the FDA regulations are considered guidance, similar to other FDA guidance. Refer to the table at the end of this document.
In protocols or contracts/agreements, GCP as adopted by the FDA may also be referred to as: Good Clinical Practice, GCP, ICH GCP as adopted by the FDA, GCP as ratified by the FDA, or GCP as described in the FDA Regulations.
FDA regulated clinical trials are those using an investigational product. The term investigational product includes drugs, biologics, devices and diagnostics when the objectives of the research involve assessing the safety or efficacy of the agent or pursuing approval from the FDA for a new indication. In some cases, dietary supplements and food additives can be considered investigational products. Any study involving an investigational drug, biologic or device is considered FDA-regulated, including IND Exempt and Non-Significant Risk determinations.
As it pertains to the conduct of clinical trials using an investigational product involving human subjects, study teams are expected to apply GCP encompassed within the applicable FDA regulations including 21 CFR 11, 50, 54, 56, 312, 812 and 814 (subpart H).
Does the National Institutes of Health (NIH) require that I follow GCP on studies for which they provide funding?
NIH encourages researchers to follow GCP as best practice for NIH funded clinical trials, but does not currently require that clinical trials comply with GCP. The NIH requires training in GCP to help ensure the safety, integrity, and quality of clinical trials.
What does it mean to follow GCP as adopted by the FDA?
To follow GCP as adopted by the FDA means to follow GCP guidelines that have a corresponding, applicable FDA regulation. As noted previously in this document, application of GCP guidelines that do not have an exact corresponding applicable FDA regulation is optional (refer to statement above from FDA regarding the purpose of FDA guidance documents). The table below includes those requirements that are unique to ICH GCP, but do not have a corresponding FDA regulation. All other GCP guidelines have an applicable FDA regulation and should be applied.
What if I am supposed to follow GCP as adopted by the FDA, but don’t?
GCP “as adopted by the FDA” means GCP guidelines that have a corresponding, applicable FDA regulation. You are required to follow GCP as adopted by the FDA if you are conducting FDA regulated research or if the clinical trial agreement requires you to follow GCP as adopted by the FDA. Failure to follow GCP as adopted by FDA in these cases could result in findings of noncompliance, a report to the FDA, or a breach of contract, which could put the UW at risk of monetary penalties and which may be assessed to your department or lab, among other results.
If I am conducting and FDA regulated trial, do I need to follow GCP criteria that are not within FDA Regulations?
The FDA recommends following GCP guidelines within its guidance documents. In the Introduction of the FDA’s guidance on GCP, it states, “In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.”
UW-Madison practices for citing findings are aligned with FDA expectations as noted above. As such, if a study is not FDA regulated and there is no contract requiring application of ICH GCP criteria that have not been adopted by FDA, study teams will not be cited for not following ICH GCP guidelines that are unique to ICH GCP (i.e., those that do not appear in FDA regulations) unless the lack of doing so presents a more significant potential impact on subject safety or data integrity.
What if my contract or protocol states that ICH GCP will be followed? How does ICH GCP compare to FDA regulations?
The ICH GCP guidelines generally agree with the FDA regulations, but there are a few areas in which ICH GCP guidelines have requirements that go beyond the FDA regulations and are considered unique to ICH GCP.
By stating or agreeing to follow ICH GCP guidelines, you are committing to following guidelines in addition to those consistent with the FDA regulations. If your study is supposed to follow ICH GCP guidelines, but does not, you could be cited accordingly.
| FDA and/or ICH GCP Section/Reference | Description |
| ICH GCP E6 4.8.8 and 21 CFR 50.27(a) |
ICH GCP Guidance states that “prior to a subject’s participation in the trial, the written informed consent form should be signed and personally dated by the subject or by the subject’s legally acceptable representative, and by the person who conducted the informed consent discussion.”
The FDA regulations (50.27[a]) (Protection of Human Subjects 2016) only require the signature of the subject and the date the subject signed the consent form. |
| ICH GCP E6 4.8.11 and FDA reference |
ICH GCP Guidance requires that the subject or the legally acceptable representative (LAR) receive a signed and dated copy of the written informed consent form.
The FDA regulations allow subjects to receive either a signed or unsigned copy, as long as it is the IRB-approved version of the consent form. |
| ICH GCP E6 4.3 |
ICH GCP Guidelines require a qualified physician (or dentist when appropriate) investigator (or sub-investigator) to be responsible for trial-related medical (or dental) decisions.
The FDA does not explicitly require this. |
| ICH GCP D6 4.9.0, multiple 21 CFR 312 and 812 references | The FDA regulations require records be adequate and accurate, however ICH GCP guidelines are more detailed, and require records be attributable, legible, contemporaneous, original, accurate, and complete (ALCOAC). However, the FDA Compliance Program Bioresearch Monitoring Manual 7348.811 for the inspection of Clinical Investigators and Sponsor-Investigators describes the document requirements as attributable, legible, contemporaneous, original, and accurate (ALCOA). |
| ICH GCP E6 5.5 and multiple 21 CFR 312 and 812 references | FDA regulations describe that the sponsor is responsible for the trial management, data handling, and record keeping. However, ICH 5.5.3 also requires the sponsor to ensure that the electronic trial data handling and/or remote electronic trial data systems conform to the sponsor’s established requirements for completeness, accuracy, reliability, and are validated and that sponsors maintain SOPs for these systems. |
| ICH GCP E6 8.1 | ICH GCP Guidelines describe the documents that are considered Essential Documents and conditions under which they should be stored and retained. Specifically, ICH GCP describes:
|
| ICH GCP E6 Section 8 | The use of a Delegation of Authority Log is described in GCP, but not explicitly in the FDA Regulations. It is, however, described in the FDA Compliance Program Bioresearch Monitoring Manual 7348.811 followed by the FDA inspectors. |
| Applicable to Sponsor-Investigators | |
| ICH GCP 5.2.2 and 21 CFR 312.52 | Both the ICH GCP Guidelines and federal regulations allow the sponsor to delegate (in writing) responsibilities to a contract research organization (CRO), but ICH GCP also requires that the sponsor ensure oversight even if duties are subcontracted to a CRO. FDA regulations state that if all obligations are transferred, a general statement that all obligations have been transferred is acceptable. ICH makes no such mention of a general statement being acceptable. |
| ICH GCP E6 5.0 | ICH GCP Guidelines state that the sponsor should implement a system to manage quality throughout all stages of the trial process, and the quality management system should use a risk-based approach, including identification of study risks to determine which may safely be omitted from continual monitoring. |
| ICH GCP E6 5.18.3 | ICH GCP Guidance states that the sponsor should:
The FDA Regulations only state that the sponsor is responsible for ensuring proper monitoring of the investigation. |
| ICH GCP E6 5.18.7 and 21 CFR 312.50 |
ICH GCP Guidance requires sponsors to develop a monitoring plan tailored to the specific human subject protection and data integrity risks of the trial.
The FDA Regulations only state that the sponsor is responsible for ensuring proper monitoring of the investigation. |
| ICH GCP E6 5.20.1 | ICH GCP Guidance requires the sponsor to take prompt action to secure compliance when noncompliance with the protocol, SOPs, GCP, and/or applicable regulatory requirements is discovered. ICH E6 further requires the sponsor to perform a root cause analysis and implement an appropriate corrective and preventive action depending on the significance of the noncompliance. The FDA Regulations do not explicitly describes these tasks. |
| ICH GCP E6 5.20.2 |
ICH GCP Guidance states that if the sponsor identifies continuing and/or serious noncompliance, the sponsor should end the investigator’s/institution’s participation in the clinical trial and notify regulatory Authorities.
Also described in 7348.811 Bioresearch Monitoring Manual followed by the FDA inspectors, Section M. Monitoring |